微生物是指肉眼看不到的微小生物,它们包括真菌、细菌、藻类和一些真核原生生物等。自然环境中微生物无处不在,它们不但存在我们生活的环境(水、空气、食品)中,也存在于几千米深的海水、地壳下三千米深的岩土和海拔八千八百米珠穆朗玛峰的土壤中。实际上,地球上微生物的生物总量相当于动物和植物生物量的总和;就是在我们人类的身体里也有大量的微生物,它们的总重量在1.5到2公斤,其细胞总数是人体自身细胞数量的十倍。因此,近些年来有一个说法,就是“人体其实是一个微生物综合体”。而实际生活中,人类也是离不开微生物的。我们生活在一个充满微生物的环境中,但不幸的是我们对它们的了解甚少,因为可以在实验室培养的微生物种类,数量不到地球上微生物总数的0.1%,因此,研究微生物的功能需要现代的分子生物学和基因组学技术。自从列文虎克发明第一台显微镜“看见”微生物,人们不断的揭示和认识越来越多的微生物,甚至能做到利用微生物来服务人类。那么到底环境中都有些什么微生物?而它们又都做了什么来改变所处的环境呢?借助新一代的生物技术,研究者对微生物的了解逐步深入。

图1 环境中微生物无处不在(水体、土壤、口腔、肠道等)
当前,通过什么方法能够深入研究和分析不同环境中众多微生物的种类和功能呢?其最佳方法主要是宏基因组学技术,或称元基因组学技术。宏基因组是指不同生境中全部微生物遗传物质的总和,宏基因组学就是采用分子生物学和基因组学方法研究微生物的菌群结构和生物功能的新学科;而宏基因组学技术则是指采用高通量DNA测序和生物信息学方法研究微生物的菌群结构和功能的技术。宏基因组学技术主要分两个不同方式,第一种方式是微生物DNA条形码目标基因的扩增和测序,主要用于微生物群落结构分析,不能进行功能基因分析。针对细菌和古细菌的DNA条形码是16S RNA基因,针对真菌的是18S RNA基因和ITS序列,针对真核原生生物的是线粒体基因组CO1基因;只针对一个或两个目标基因的扩增和测序,可使测序成本低。宏基因组学技术的第二种方式是对微生物宏基因组进行全基因组测序。这种方式DNA测序比较简单,但测序量大,后期的生物信息分析较为复杂,可获得的微生物菌群结构比较准确,同时还可以进行微生物的基因功能分析。这两种方式的流程基本一样,主要包括,1)提取环境中的基因组DNA,2)基因组DNA分子文库的构建及高通量测序,3)高通量DNA序列数据的生物信息学分析。
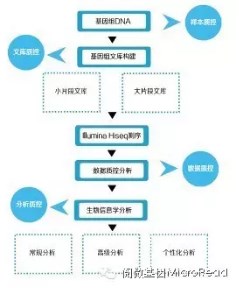
图2宏基因组测序基本技术路线
然而我们如何来对高通量测序得到的数据进行分析?能得到哪些结果呢?针对目前论文比较常见并得到认可的方法,其概括如下:1)DNA条形码序列是微生物系统发育和分类鉴定的常用指标,测序获得DNA条形码高变区序列,使用Mothur、QIIME等分析流程得到OTU聚类结果,然后将OTU序列比对GreenGenes、Silva、RDP等数据库进行物种注释,采用alpha多样性及beta多样性分析来反映环境中微生物的多样性及种群结构等信息。2)宏基因组分析是将高通量测序获得的数据采用SOAP denovo、MetaVelvet、MetaIDBA、MetAMOS等拼接软件进行基因组序列组装,然后采用MetaGeneMark、Orphelia等软件进行基因预测,获得的基因序列比对NCBI-NR/NT数据库进行物种注释,比对COG、GO、KEGG、eggNOG、CAZy等数据库进行基因功能注释,采用MEGAN、ShotgunFunctionalizeR等多样性分析软件可以揭示样本之间的物种多样性与丰度差异、基因集组成及其功能和参与的代谢调控网络,探求微生物与环境之间的关系,发掘具有特定功能的基因。这里需要特别提一下细菌基因组测序,将高通量测序获得的数据采用SOAP denovo、SPAdes、Velvet、ABySS、ALLPATHS等拼接软件进行基因组序列组装,然后采用Glimmer、GeneMarkS、Prodigal等软件进行基因的预测,获得的基因序列比对COG、GO、KEGG等数据库进行功能注释,最终分析得到此细菌基因组的序列信息及基因功能注释等信息。
了解了什么是宏基因组测序,也清楚了怎么进行实验和分析,我们接着来看一看案例,以便更好的掌握这个技术。首先探讨牙周炎跟微生物的关系。
以往研究通过培养和非培养手段发现了一些在牙周炎患者口腔中广泛存在的细菌,如Porphyromonasgingivalis、Treponema denticola、Tannerella forsythia等,但是早期研究主要集中于已知致病细菌的代谢途径,而对于口腔中其他细菌的功能研究则极少涉及。现代研究应用宏基因组测序技术,针对健康人群和牙周炎患者口腔微生物的功能差异进行研究。
研究发现,健康人群和牙周炎患者口腔微生物丰度不同,Bacteroidetes在牙周炎口腔丰度最高,Actinobacteria和Proteobacteria在健康口腔丰度显著增加。
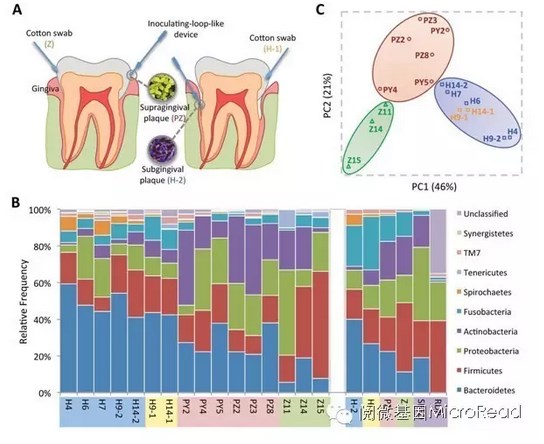
图3健康和病人牙周样本采集、构成和细菌群落的聚类关系
应用宏基因组测序技术也证实了早期的研究成果——P. gingivalis与牙周炎相关,在牙周炎样本和健康样本之间具有明显差异。
为了研究牙周炎样本与健康口腔样本间功能差异,将其序列与KEGG数据库进行比对,发现二者在膜转运、信号转导和细胞运动性相关基因的数量上具有显著差异(Figure 4)。绝大多数与细节趋化性相关的基因在牙周炎样本 (H-2)中数量显著增加 (p<0.001),如甲基受体趋化性蛋白 (MCP)是趋化组氨酸蛋白激酶(CheA)的2-7倍,CheW、CheY、CheB和CheR也显著增加。但是,并非所有的趋化性均由MCP调控,磷转运系统 (PTS) 中的Pts I会抑制CheA,研究结果显示,可能是由于H-2中的PTS基因数量减少,使得对CheA的抑制作用减弱,从而增强了细菌的趋化性。
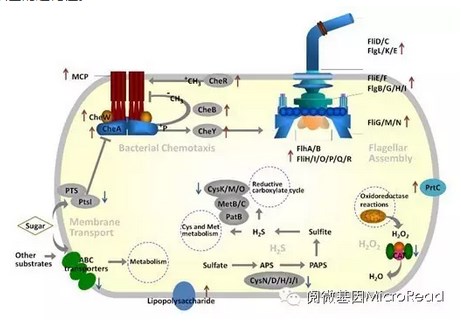
图4牙周炎相关微生物功能变化的原理图概述
接着我们来探讨大家经常说的耐药性,新闻中总说:“不要滥用抗生素,容易产生耐药性”,耐药性到底是怎么回事?人类肠道寄居着大量微生物,这些微生物群有许多耐药基因,即使短期使用抗生素,耐药菌群也会在人体内存活多年。而且,不同地区的微生物群耐药基因型有差别,中国人和丹麦人有133个耐药基因型,西班牙人有128个耐药基因型。仔细一想,人各有不同,这个也正常。

图5为了比较不同地区的基因多样性,根据测序覆盖度计算了每个基因的相对丰度。中国人、丹麦人、西班牙人耐药基因占肠道基因的0.94,0.44,089%。
看到这里,相信大家都明白了宏基因组测序技术能做些什么了,希望对大家有助益,让您的研究时尚、有效起来。阅微基因也会持续跟进各位大牛的步伐,提供更多更好的服务。如果有了新的学习心得,再跟大家汇报!






