



这场300+老师共同探讨的10x Genomics 直播课都说了啥?

流式细胞分选是经典的方法,只要有相应抗体就可以完成分选。但在实际细胞分离当中,由于流式分选价格相对昂贵或设备限制,实验者会考虑采取其他方法分离细胞,试剂盒分选就是其中之一。
与流式分选相比,试剂盒也能够高效、简便地完成目的细胞分选,极大地提升了可操作性和节省时间成本。目前市面上推荐的试剂盒通常是磁珠+特异性抗体结合的方法,例如美天旎品牌的磁珠分选,其技术水平成熟,已可应用于临床。例如,从人或小鼠组织中已可分选出纯度高达95%的naive CD4+ T细胞,且分选出的细胞活性高,活化状态及功能不受影响。
当然,利用试剂盒分选的前提还是必须有适于分选目标细胞类型的产品,并进一步确认其分选的效率、时间等因素。
值得注意的是,下面列出几种其它细胞分离方法不能替代的情况,此时必须进行流式的细胞分选:
■ 要求目标细胞群有极高的纯度(95%~100%);
■ 需要分离低密度表面受体/抗原的细胞(弱阳性细胞);
■ 需要根据表面受体密度的差别来分离不同细胞(根据免疫表型的分选,抗体亲和力进化等);
■ 需要依据多色荧光标记来分选细胞;
■ 根据某些细胞内部标志,如DNA含量、胞内抗原等分离细胞;
■ 多孔板分选:如单克隆分选,在96孔板每个孔中精确分选进1个细胞;
■ 需要分选极低含量的细胞(如0.001%或更低),流式可以分选出百万分之一的稀有细胞。
Q3: 对于肿瘤组织来说,一个比较大的肿瘤组织可能有几千万个细胞。怎么收集其中的肿瘤细胞呢?需要用什么marker先分选吗?
前期分选肿瘤细胞的难度较大,一般肿瘤组织中健康细胞比例要远低于肿瘤细胞,可以直接取肿瘤组织,在测序结束后利用基因表达的模式对不同类型的肿瘤细胞和健康细胞进行筛选;如果想先收集肿瘤细胞,则可考虑流式方法甚或活细胞显微切割技术,要在确定该癌种肿瘤细胞有明确marker的前提进行,同时这种marker还应具有相应的抗体。
Q4: 用类似RNAlater固定以后再分离能不能做10x单细胞测序?
不可以。RNAlater虽然常用于防止RNA降解,但其含有能变性裂解细胞的成分,且浓度比细胞质液高得多,会造成细胞破裂。而10x单细胞测序要求细胞是活的,应使用专门的组织保存液来保存组织,保证细胞的活性。
Q5: 能直接拿培养的细胞做吗?
可以的。培养的细胞系是10x单细胞测序中比较常见的样本类型。近两年已有不少发表的文献中直接以培养的细胞系进行单细胞测序的。[1]
Q6: 如果一个样本分选完细胞只有1~2万个细胞,如何来收集细胞并调节到合适浓度呢?简单的离心可以吗?
可以离心。但是离心的速度需要根据细胞量和细胞大小进行调整,以确保细胞完整性和活性。
Q7: 如果一次上机得到的细胞数超过了15,000,已经是根据常规参数过滤了一遍的,其他的质控参数都很好,还有哪些参数可以设置或者怎么设置来过滤细胞呢?
一般来说,10x Genomics捕获到每个样本的细胞最多为1万,细胞数量再进一步增加会使油滴中多细胞概率大大上升,影响到后续分析,但是确实不排除可以尝试更多细胞捕获的做法。如问题所示,如果这15,000个细胞已经用基因数量、转录本数量和线粒体基因比例等参数都过滤完了。那么说明这些细胞是合格的,不需要进一步过滤。可以进入后续分析,例如细胞亚群聚类和差异基因分析等。
问题集合2:关于细胞量
Q1: 为了获得更多的细胞量,要制备更多的样品是吗?如果要达到百万级别的细胞量,那就是需要超过100个样本是吗?
一般来说,为了获得更多的细胞量,需要更多的样品来建库。因为在建库中每个样品捕获到的细胞量是有限制的。但同一个来源样本可以分成多个样品进行建库测序,因此百万级的细胞可能不需要100个不同的样本,但确实需要进行至少百余次的建库。或者选择其他更大通量的单细胞测序方法。
Q2: 为什么一个样本中只提取6K-8K个细胞来进行测序?是目前技术限制吗?有些组织可能会含有上百万个细胞,只提取其中少量细胞进行测序是不是覆盖面不够?
上文提到,10x Genomics捕获到每个样本的细胞是最多为1万,现在10x的技术,细胞数量再进一步增加会使油滴中多细胞的概率大大上升,影响到后续数据质量。根据我们的经验和文献当中单个样本捕获到的细胞量综合考虑,一个样本捕获6K-8K个细胞是成功率高且最为稳妥的方法。
6K-8K细胞的覆盖率确实是有限的,然而我们可以通过将一个样本分成多个样品进行建库来增加测序的总细胞数量。而测序总细胞量越多,可分析到的内容就越全面。但同时,经费成本也成倍增加。所以需要视研究目的来选择方案。比如目的是进行组织中单细胞表达图谱构建,想尽可能地覆盖到更多细胞类型时,细胞量必须多一些,则每个样本可以制备多个建库样品。而如果是想比较不同疾病间某类型细胞基因表达差异时,如果该细胞在组织中的丰度尚可,则6K-8K个细胞便能满足。
Q3: 请问在鉴定罕见细胞亚群时,需要多少细胞才能被鉴定从而聚类呢?
如果是想讨论不同亚型细胞的功能、发育和分化和能否得到精确的细胞亚型则需要进行细胞数量的计算。主要有两种计算方法:
方法1:依照粗略的计算公式,计算测序所需细胞量。计算公式为:
P(d)= 1-(1-s)n
P(d):检出力
s:目标细胞亚群比例(一般为最小比例细胞亚群,也称为罕见亚群)
n:需要被测序的细胞数量
此公式换算后为:n=lg (1- P(d))/lg(1-s)
依照此公式,当组织中或者细胞悬液中目的细胞亚群比例为1%,检出力达到0.9时,则需要250个细胞被测序。当目的细胞亚群比例为1%,检出力达到1时,则需要500个细胞被测序。需要注意的是,这种粗略计算方法没有考虑到降低假阳和假阴性错误率,以及消除随机误差所需要的细胞重复数,一般所需的细胞重复为2-5次重复。
方法2:
第二种细胞测序数量估算方法和第一种估算方法相比,除了考虑最小频率细胞亚群、检出力,还考虑了可能的细胞亚群数量和最小频率细胞的最低细胞检出量。例如当所有细胞可聚类成10个亚群,最小比例细胞亚群比例为3%(0.03),检出力达到0.9且最小比例细胞亚群至少有50个细胞被检测到,需要的测序细胞量是2200个细胞。
针对此算法,Satija 实验室已经开发了一个网络工具评估细胞测序数量,链接:https://satijalab.org/howmanycells,在网站中可以调节参数计算测序细胞数量。
Q4: 如何粗略估计每个样品大概能获取多少个细胞数量呢?
10x Genomics的系统捕获效率最高达65%,在实际的操作中,我们会按照捕获效率50%进行上样,适当的多上一些细胞。因此如上样1万个细胞,保守估计捕获到的细胞数量约是5000个。
问题集合3:技术原理
Q1: 请问单细胞建库方法5’端和3’端捕获有什么区别?为什么VDJ通常用5‘捕获?
10x单细胞转录组建库有5‘端和3’端建库两种方式。在3’端的建库中,Gel beads的末端序列是poly(dT),用于和转录组3’端的polyA结构互补配对。而在5’端的建库中,Gel beads的末端序列是TSO序列,其末端有polyG的结构。转录本会在酶的作用下在其5‘端加上polyC的结构,由此完成配对[2]。
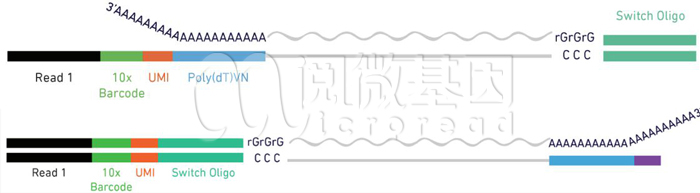
VDJ建库的捕获固定采用5’端捕获方法,目的是为了在保证序列长度较短的情况之下富集到位于5’端的多变VDJ区域,而不是位于3’端的较为保守的C区域,这段区域通常不是重点的研究方向。
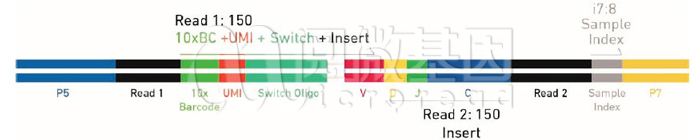
Q2: 750万种GEMs, 不同的区别是什么呢?
在10x的试剂中,每一个beads上面的barcode序列都是不一样。而在一个芯片孔中,至多检测到的细胞为1万个,至多上机细胞数在2万。750万和2万的比例是相差很大的。这样做是为了保证包裹在油滴中的细胞具有唯一的barcode。
问题集合4:生信分析
Q1: 在拟时序的分析中,什么是伪时间的开始?
细胞的分化发育是一个有方向的不可逆过程,而拟时序分析就是根据不同的细胞状态来模拟细胞分化的过程。伪时间是衡量我们感兴趣的某细胞类型在整个拟时序轨迹当中处在一种什么状态的数值,而伪时间的开始一般指在细胞轨迹的构建中,最原始状态细胞或早期细胞所在的节点位置(如下图当中的节点1或2)。
目前拟时序分析中常用的是Monocle R包,在完整的包中已建立23730个基因和301个样本库[3]。其分析的步骤是将测序的差异分析结果选取适合的 Marker 基因加载,再进行降维和排序后便可以得到拟时序轨迹,而特定细胞亚群在轨迹上的位置与根部(可能是一群祖细胞)的距离即为伪时间。自然,多个细胞亚群便可以有多个起点和轨迹。
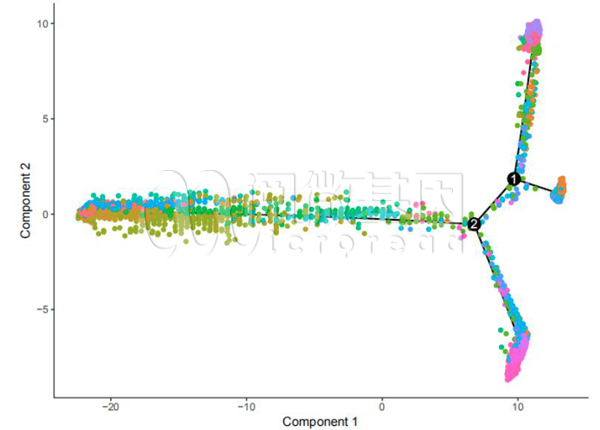
细胞拟时序分析示意图:图中每个点代表一个细胞,具有相似细胞状态的细胞被聚到一起,每个分支点代表着一个可能的细胞生物学过程决策点(该例图中有2个分支点)。
问题集合5:实验设计
Q1: 单细胞转录组测序在取样时是否可以取癌组织部分和癌旁组织,混合成一个样?还是必须分开来测,算作两个样?这样的情况目前发表的文章里面出现过吗?
样本是否能够混在一块要看具体研究目的。若目的是想比较正常组织与癌症组织的差别,那么最好分做两个样本。若因经费问题导致不同样本混在一块检测,混合比例是一个需要注意的问题;若是想看癌症的发展历程,则可用单个癌症组织,因为癌症组织中也含有正常细胞,早中晚期各个阶段的细胞。
Q2: 为了达到测序数目的要求,可以把同组的多只小鼠的某一组织混合起来作为一个样本?
为了得到足够的细胞数或平衡个体差异可以这样做,也有很多文献采取这样的方法[4]。但需要注意这种方法适用于需要控制上机样本数量,又同时需要增强此组样本细胞种类全面性的场景。
没看过瘾或想问其他问题?扫码添加阅微基因客服微信咨询,专业科研管家为您解答。




