



16S/18S rDNA包含可变区和保守区,原核微生物的16S rDNA基因长度约为1500bp,真核微生物18S rDNA基因长度约为1500-2000bp。保守区在菌种间差异不大,可反映物种间的亲缘关系,高变区具有属或种的特异性,根据物种亲缘关系不同而有一定的差异。因此16S /18S rDNA成为了国际公认的微生物系统发育和分类鉴定的指标。
传统16S/18S 扩增子测序仅针对16S/18S rDNA的单个或连续的两到三个可变区进行分析,而基于三代测序平台PacBio的全长16S/18S 扩增子测序可轻松读取微生物16S/18S rDNA全长序列,突破了二代测序读长较短的局限性,提高了菌株在种水平的分辨能力,真正精确到“种”的分类鉴定。全长扩增子测序获得全部变异区域序列信息不仅能提高物种鉴定的分辨率,还能提高样本中微生物组成鉴定的精确度,从而更加真实的还原样本中微生物的群落结构。
全长16S/18S测序常见的应用领域如下
医学领域:疾病与人体肠道、表皮微生物的关系
动物领域:肠道、瘤胃(如产甲烷菌类群)与动物健康/营养消化研究等;
农业领域:根际微生物与植物互作、农作/农药/施肥与土壤微生物群落等;
环境领域:雾霾处理、污水治理、石油降解、酸性矿水处理及海洋环境等;
特殊极端环境:深海、高原、荒漠等极端环境下的微生物类群、资源研究
技术路线
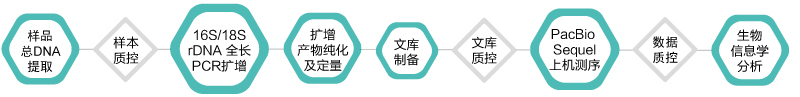
技术优势

测序策略
|
测序策略: |
PacBio Sequel 2kb 文库 |
|
建议数据量: |
≥5000 CCS reads |
收样要求
|
土壤、污泥、沉积物 |
15g 以上 |
| 粪便 | 5g以上 |
|
血液 |
10 mL |
|
DNA |
总量≥200 ng,浓度≥10ng/μL,
有明显主带,无降解,无RNA蛋白质污染,
OD A260/280:1.8 - 2.0
|
|
PCR 产物 |
电泳条带单一,大小在1-2kb之间 |
您可以得到的数据分析
生物信息分析流程
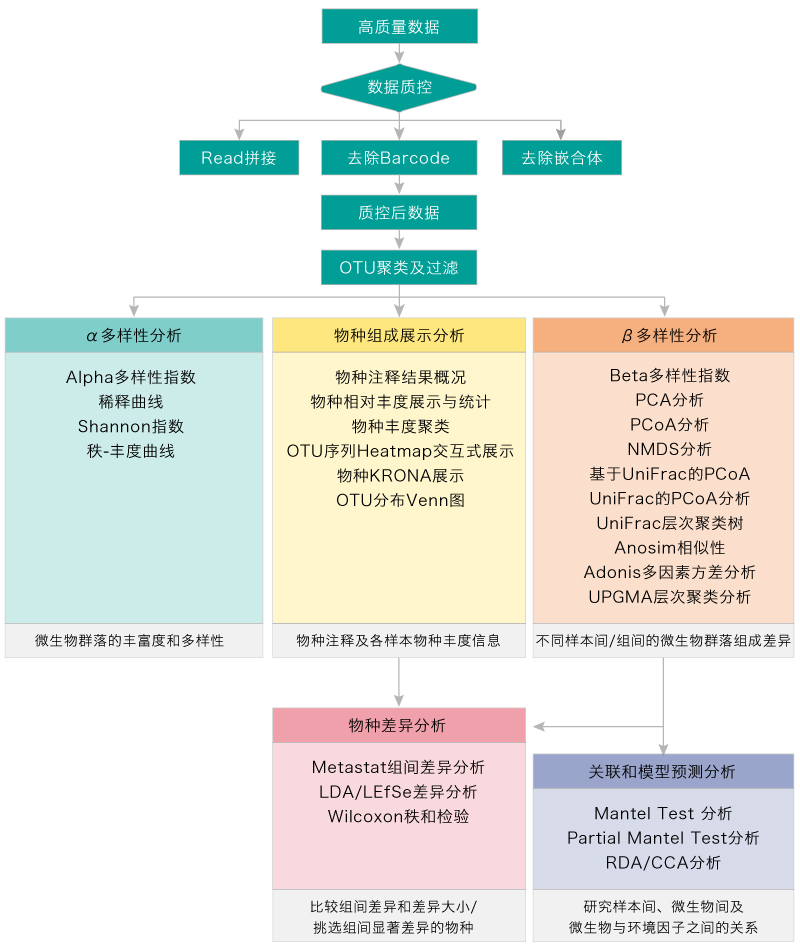
|
物种组成展示分析 |
α多样性分析 |
β多样性分析 |
关联和模型预测分析 |
组间差异分析 |
|
OTU聚类与过滤 |
Alpha多样性指数 |
Beta多样性指数 |
Mantel Test分析 |
Metastat 组间差异分析 |
|
物种注释结果概况 |
稀释曲线 |
PCA分析 |
RDA/CCA分析 |
LDA/LEfSe 差异分析 |
|
物种相对丰度 展示与统计 |
Shannon指数 |
PCoA分析 |
Partial Mantel Test 分析 |
ANOSIM 相似性分析 |
| 物种丰度聚类 |
秩-丰度曲线 |
NMDS分析 |
|
Wilcoxon 秩和检验 |
|
OTU序列Heatmap 交互式展示 |
|
基于UniFrac的 PCoA分析 |
|
|
|
物种KRONA展示 |
|
UniFrac层次聚类树 |
|
|
|
OTU分布Venn图 |
|
Anosim相似性 |
|
|
|
|
|
Adonis多因素 方差分析 |
|
|
|
|
|
UPGMA层次聚类 分析 |
|
|
案例解析
模拟菌群(已知单菌混合)和湖水样本的全长16S测序与V4区域测序对比
在过去的很长时间里,基于二代测序平台的扩增子测序是研究微生物多样性很重要且常用的方法。随着三代测序的出现,全长16S/18S测序的方法可以大大提升环境中微生物物种组成的分辨率,提供更加详细的信息。
本研究中,作者利用PacBio SMRT RSII 平台对整段16S rDNA进行测序,得出的全长基因序列称为PhyloTags,同时也利用Illumina Miseq 平台对16S rDNA的V4区域测序,得出的序列被称为iTags。首先对含有23株细菌和3株古菌(有参考基因组)的模拟群落的两种测序结果进行对比,并以Pacbio鸟枪法测序(无扩增偏差)的结果作菌群结构比较的基准。结果显示:模拟群落(n=5)的高质量PhyloTags数据成功被分成22个OTUs,每个OTU内的任意两条序列相似度高于97%。各个OTU中质量最好的PhyloTags与16s rDNA参照序列的一致性达到99.5%。另外,5个PhyloTags技术重复的相关性非常好,两两相关系数均达到0.96以上,并与作为基准的鸟枪法测序具有强相关性(0.77)。而iTags结果在菌群构成分析上与鸟枪测序整体相关性更高(0.89),显示PCR扩增短序列的偏差性较低,但iTags数据存在约0.05%的污染序列,且对部分特定菌种的估计偏差较大。
Singer E et al.,“High-resolution phylogenetic microbial community profiling”The ISMEThe ISME journal(2016)DOI:10.1038/ismej.2015.249(Impact factor:9.664)
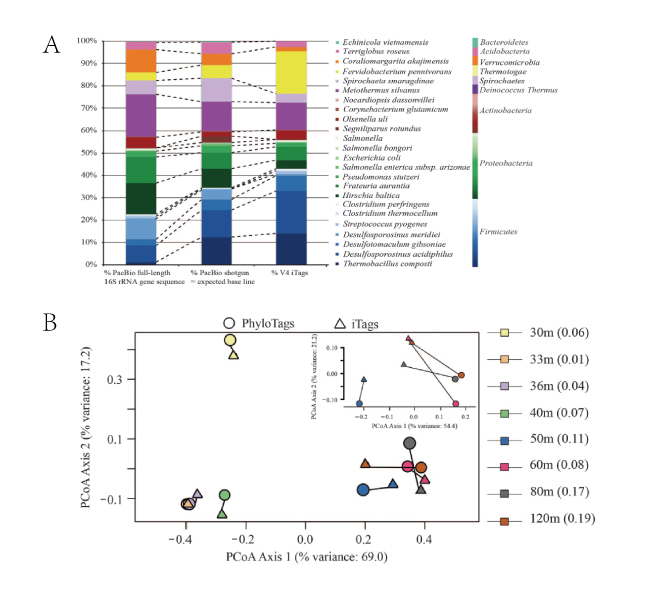
A. 基于PhyloTags(n=5),PacBio shotgun(基准)和V4 iTags的菌群分布柱状图。丰度非常低的Nocardiopsis dassonvillei菌只在PacBio shotgun(基准组)组中出现,模拟菌落外的OTUs(预测错误)只在iTags组中出现。
B. Sakinaw湖不同深度水样中微生物群落的主坐标分析(PCoA)图:圆圈代表PhyloTags,三角形代表iTags,相同深度的PhyloTags和iTags用线连接表示。在50-120m的湖水深度样本中,两种测序方式在差异明显。
小鼠肠道微生物16S全长测序与短读长测序对比
随着医药和健康产业的快速发展,对环境和临床样本中微生物群落的研究要求更快更准。二代测序在微生物领域的普及使得要求得以实现,但由于其读长短,测定区域有限的特点,在微生物种群中属的分类上并不是那么准确。为了解决这个问题,本文利用Nanopore测序平台对小鼠粪便中的微生物群落进行了16S rDNA全长测序和短读长测序的对比和研究。
研究者从两只小鼠(A、B)的结肠粪便中提取DNA,同时利用Nanopore MinION 对16S rDNA全长覆盖V1–V9 进行测序,和利用Illumina MiSeq (PE250)对16S rDNA的 V3-V4 可变区进行测序。实验结果显示,Illumina MiSeq 测序仪产出数据平均长度约为447 bp,Nanopore MinION 测序仪产出的数据读长分布集中在很窄的区域,平均值为1,393 bp,接近 16S rDNA全长(1,550 bp)。两种方法一致检出8个细菌门和13个纲。在统计学上 Nanopore 与 Illumina 在宏基因组的门和纲水平分类上高度相似(R2>0.83),而 Nanopore 在种水平有较好的分辨率。通过系统发生学分析确定了分布在13个属的16个不同的种。其中拟杆菌属(Bacteroides)的两种菌(Bcidifacies,B.ovatus)能同时被 Nanopore MinION 和 Illumina 两种平台区分测定,而双歧杆菌属(Bifidobacterium)的两种菌(B.animalis,和B.psudolongum)只被Nanopore MinION 区分检出。
综上所述,Nanopore 是第三代测序技术中除了 PacBio SMRT 之外另一种测序技术,虽然目前Nanopore MinION 有比较高的原始错误率,但其体积小、使用成本低、文库构建时间短(<3h)、可实时测序,在微生物群体的快速分类鉴定中应用前景广阔。
Shin J et al., “Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing”,Scientific reports(2016) DOI: 10.1038/srep29681 (Impact factor:9.664)
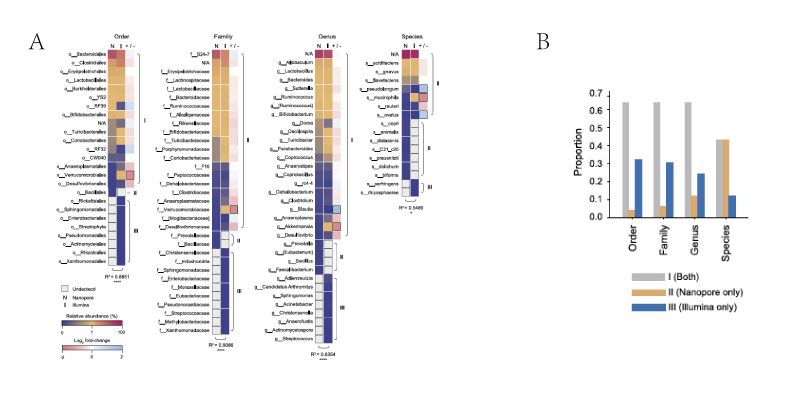
A. 从目、科、属、种水平上比较两种测序方法得到的微生物群落相对丰度热图。由蓝到红丰度增高;灰色:未检出。N:Nanopore;I:Illumina;+/−:log2变换,黑框标记表示有明显丰度差异。
B. 在目、科、属、种水平上三种检出情况的比例。灰色:两种方法共同检出,黄色:只Nanopore MinION检出,蓝色:只 Illumina Miseq检出。
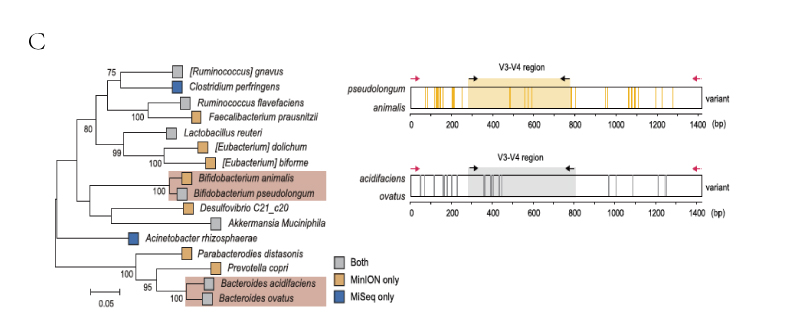
C(左). 极大似然种系发生树。底色为红色的框表示从属中区分出的种。
C(右):拟杆菌属的 Bacteroides ovatus和 双歧杆菌属的Bifidobacterium animalis 16S rRNA序列变量(variants)分析, 说明Bifidobacterium animalis 在V3-V4区的变量较少,因此仅在全长分析时可被区分出。
研究趋势与研究热点
1.人体或动物微生态,如肠道菌群,皮肤菌群的结构和多样性的探究
2.自然环境微生态,如作物根系或受污染等土壤微生物,海洋、污水、污泥等环境微生物菌落结构和多样性探究
3.工业环境微生态,如食品发酵液,工业废水、污泥处理器中微生物的多样性,以及产甲烷菌、硝化/反硝化
4.细菌的功能基因研究
5.除上述环境外的特定环境中微生物分布及多样性调查
6.环境核心微生物/特定关注微生物在不同处理间的差异展示
7.在食品或药品的微生物安全检测中也有所应用
客户常见问题
全长16S/18S测序的优势和劣势是什么?
全长扩增子测序可以得到更长的读长和更全的扩增区域,基于此,与传统基于二代平台上的扩增子测序相比,可以得到更细致的物种分类和更多物种注释信息。当然,全长16S/18S的缺点也很明显,就是成本比较高,所以该如何选择,需要具体看研究目的了。
鉴于文献,若需要鉴定到种水平,且保证OTUs数量能够覆盖样品中97%以上的微生物,各类型样品所需数据量如下:肠道、粪便样本:3000-4000条reads;水体样本:3000-4000条reads;土壤样本:4000-5000条reads。
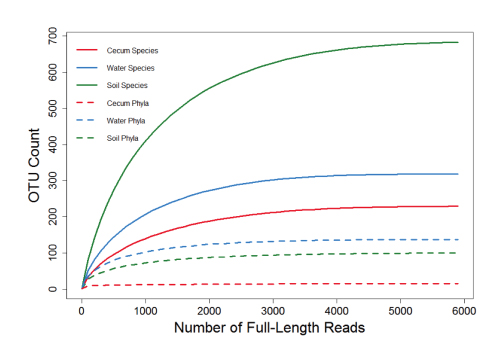
横坐标为全长CCS的数目,纵坐标为OUT数目。可以看出,各类样本在测序量达到5000CCS以上,OUT已经不再增加,说明5000 CCS基本能满足各类样本的研究需要。
Bowman et al., "Analysis of full-length metagenomic 16S genes by SMRT sequencing" Chem, 2013, 4: C2.


